 全国服务热线
全国服务热线189-4472-1685

文章编号: 1000-3851(2012)02 -0016-10
编辑:弗艾博浆纸科技发展(广东)有限公司
刘杰*1,白艳霞1,田宇黎1,黄翔宇2,王春华1,梁节英1
(1.北京化工大学碳纤维及功能高分子教育部重点实验室,北京100029; 2. 上海石油化工股份有限公司,上海200540)
摘要:采用新型电化学 表面处理设备,以10%(质量分数)NH4HCO3溶液为电解质,对12K PAN基碳纤维进行连续化的表面处理,探索了在提高碳纤维/树脂复合材料层间剪切强度的同时降低碳纤维本征拉伸强度损失的结构变化特征及规律。利用SEM、XRD、XPS、Raman等方法研究了改性前后碳纤维表面的物理和化学状态、晶体尺寸和表面有序度。结果显示,在适当的条件下,碳纤维/树脂复合材料的层间剪切强度提高了23. 3%,同时碳纤维的拉伸强度仅损失了4. 4%。碳纤维/树脂复合材料层间剪切强度的提高是碳纤维表面粗糙度和表面含氧、含氮官能团共同作用的结果。表面处理后碳纤维石墨网片层尺寸减小了6. 3%~27.6%,微晶尺寸相应减小;适度的氧化刻蚀使碳纤维表面有序度略有提高,并且产生新的活性点;以上两种作用减小了碳纤维的本征拉伸强度的损失量。
关键词: PAN 基碳纤维; 电化学氧化法;拉伸强度; 层间剪切强度;力学性能; 表面官能团
中图分类号: TB332; TQ34 文献标志码: A
作为结构型复合材料,人们更关注的是其力学性能能否有效发挥。而制约碳纤维/树脂复合材料宏观力学性能的关键,就在于碳纤维与基体树脂间形成的界面,它是复合材料极为重要的微结构,其结构与性能将直接影响复合材料的性能。研究表明,未经表面处理的碳纤维比表面积小,表面活性低,导致其与基体树脂的结合性能差,最终影响了复合材料力学性能的发挥[1]。而表面处理在对碳纤维进行表面刻蚀的同时可以在碳纤维表面引人大量
的含氧和含氮官能团,这既提高了碳纤维的比表面积,又增加了碳纤维的表面活性,非常有利于碳纤维/树脂复合材料性能的改善[2]。据报道,对碳纤维进行表面处理的方法有很多种,如电化学法、等离子体法、气/液相氧化法、涂层法等。其中电化学氧化方法[0]因其可连续生产、简单易操作、处理条件温和易于控制,在工业上得到了广泛应用。但是,该方法存在以损伤碳纤维拉伸强度为代价来提高碳纤维表面性能的弊端。因此,如何在提高层间剪切强度(ILSS)的同时,降低碳纤维本征拉伸强度(σT)的损失,以最大限度地发挥碳纤维在复合材料中的作用,进而提高复合材料的性能是该领域研究的新目标。
前人的研究多集中在对1K、3K等小丝束碳纤维的处理过程及处理结果上[4],而随着碳纤维工业的发展,大丝束碳纤维相对于小丝束碳纤维有着越来越广阔的应用空间和发展前景,所以探索大丝束碳纤维的表面处理工艺参数及处理效果有着更为突出的意义。总结前人的研究可知,电化学表面处理后碳纤维表面活性官能团的增加、表面粗糙度的提高及弱层去除的共同贡献使层间剪切强度得到提高;而碳纤维拉伸强度的降低主要源于表面处理过程中深度氧化刻蚀作用在碳纤维表面引入了新的缺陷,这些缺陷来自于外层有序晶体层剥落后露出的内部无序结构在纤维本体上增加的薄弱点[6-8]。
因此,本文中尝试通过研究电化学改性中各项技术参数,探索合适的改性条件,控制碳纤维表面活性官能团含量、表面刻蚀程度与晶粒尺寸的变化,使碳纤维经过电化学改性后,复合材料层间剪切强度得到提高的同时,降低碳纤维拉伸强度的损失量;并通过Raman、XRD、SEM、XPS等手段解析碳纤维表面处理后层间剪切强度和拉伸强度变化的机制。
1实验
1.1 原料
PAN基碳纤维:由英国Courtaulds 12K原丝生产的碳纤维;碳酸氢铵:分析纯;环氧618树脂:江苏星辰化工有限公司树脂厂生产;三乙烯四胺;分析纯,北京化学试剂公司生产。
1.2电化学氧化处理
采用新型电化学表面处理设备,减少了传动辊的用量,通过实现电解液的循环增加了电解槽的传质效果。以0.5mol/L碳酸氢铵作为电解质溶液,电流密度为0. 15 mA/cm2,电解温度30C,电解时间为90s、120s、150s,对碳纤维进行连续化电化学氧化处理;用循环蒸馏水清洗,除去纤维表面残余的电解质;再经100 °C干燥上胶收丝。
1.3性能测试
1.3.1 碳纤维拉伸强度测试根据GB 3362-82,将丙酮、环氧树脂、三乙烯四胺以质量比20 : 10 : 1配成基体树脂溶液,将碳纤维浸泡于其中2~3 min,取出后缠绕于不锈钢成型框架.上,并于120°C固化2h,制成长度为200 mm的测试样条,并剔除具有明显折弯点或悬挂有胶液珠的残品样条。测试前将样条两端分别固定在纸质夹片中;测试采用美国装配的INSTRON-5567型万能材料实验机,加载速度为10 mm/min,夹头间距为150mm;测试结果取10个有效数据的平均值;以上过程均符合GB 3362- 82标准。
1.3.2 层间剪切强度(ILSS)测试
采用GB3357-82记载的方法进行碳纤维/树脂复合材料层间剪切强度测试。测试样品选用环氧618型树脂及三乙烯四胺固化体系。树脂及固化剂配比为10:1,二者混合后充分搅拌,并快速均匀地涂于碳纤维的表面,上,尽量使得碳纤维与树脂充分浸润。将涂覆好树脂的碳纤维放入模具中压制成型,并于120 °C固化2 h,取出样条后使用500 μm耐水砂纸打磨除去飞边,按标准要求尺寸(跨厚比.5:1)制成CFRP测试样条。测试仪器为INSTRON-5567型万能材料实验机,加载速度2 mm/min。每种试样测试结果取10个有效数据的平均值。
1.3.3 性能表征及形貌观察
使用Rigaku D/max2500 VB2+ /PC型多功能粉末多晶X射线衍射仪分析碳纤维改性前后晶体结构的变化。
使用英国Renishaw公司生产的RM2000型显微共焦拉曼光谱仪进行表征,激光器波长为514.5nm(氩离子),使用20倍物镜,光斑直径5 μm,扫描时间30s,累加次数10次。采用日本HitachiS-4700型发射扫描电子显微镜观察样品表面形貌,样品真空镀金2~3 min。
采用英国VG公司生产的Therm VG ESCAL-AB250型X射线光电子能谱仪,分析碳纤维改性前后表面化学状态的变化。射线源为MgKa(1253. 6eV),功率为250 W(12.5 kV X 20 mA)。
2结果与讨论
碳纤维及碳纤维/树脂复合材料力学性能测试结果见表1。可以看出,改性后碳纤维的拉伸强度均低于未改性碳纤维,但降低幅度均在10%以内,最小降低幅度仅为4.4%,较传统碳纤维改性后损失15%左右的强度值要小,可见通过适当调控制备参数可以在大幅提高碳纤维复合材料层间剪切强度.的同时控制碳纤维拉伸强度降低幅度<5%。随着改性时间的增加碳纤维的拉伸强度的损失量逐渐减小,强度值呈上升趋势;碳纤维/树脂复合材料的层间剪切强度均有显著的增加,但当电解时间为120s时,层间剪切强度略有下降,在改性时间增加到150s时又有所回升。氧化时间的增加相当于在同等氧化水平下,氧化刻蚀程度的加深。在较短的改性时间下,电化学氧化刻蚀首先剥离碳纤维最外层的表面,这层表面主要包含一些弱层结构,随着氧化刻蚀程度的加深,刻蚀进一步向内部深人,使得处于次外层的缺陷暴露成为表面缺陷,碳纤维的拉伸强度出现较大损失;当电解程度进一步增加时,暴露出的缺陷尖锐的部位在电化学改性的尖端效应下被钝化,减少了应力集中,碳纤维的拉伸强度随之略有回升。150s改性后碳纤维的拉伸强度仍高于120s改性,说明150 s改性时间仍在可以控制碳纤维拉伸强度的程度范围内;也可以预计当电解时间更进一步增长后,碳纤维的拉伸强度必然会因为更多的缺陷被暴露以及氧化刻蚀产生的新的缺陷而导致更多的损失。由于工业改性的改性时间大部分在1.5~3. 0 min以内,并在可能的情况下尽可能缩短改性时间,因此,可以不考虑由于改性时间的增加带来的更大的强度损失。此外,改性后碳纤维/树脂复合材料层间剪切强度大幅度增加,这归因于表面弱层被刻蚀剥离以及表面极性官能团的增加。表面含氧官能团理应随着电解时间的增加而增加,层间剪切强度也应一直处于增加的趋势,但在120s改性后复合材料的层间剪切强度反而处于一个低谷,这有可能是由于碳纤维表面形貌和含氮官能团发生变化所致。
2.1碳纤维表面的XRD分析
电化学表面处理会改变碳纤维表面晶态结构和表面形貌,进而改变碳纤维的表面物理状态。由XRD研究T-0、T-90、T-120及T-150(T-90、T-120及T-150的电解停留时间分别为.90s、120s和150s,其拉伸强度与未改性碳纤维T-0相比降幅分别为9. 0%、4. 9%和4.4%,层间剪切强度增幅分别为23. 2%、17. 7%和23.2%)的晶粒尺寸的变化。碳纤维表面石墨网层平面尺寸L。及层面堆积厚度L。可按照Scherrer公式分别由(100)晶面、(002)晶面衍射数据计算:
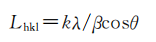
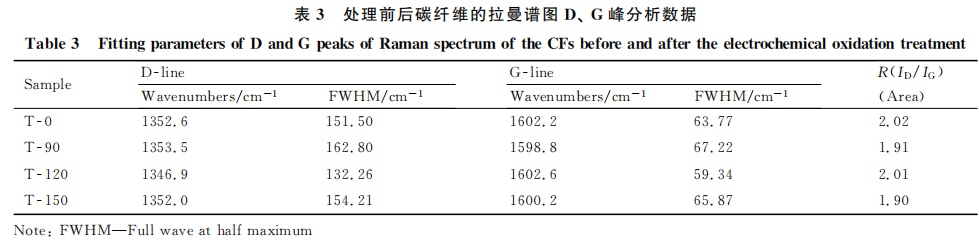
式中,k为形状因子,一般取0.89[9]。计算结果列人表2。可见,碳纤维经过电化学氧化处理后,T-90微晶的厚度Lc和宽度La均减小。微晶尺寸减小可以减缓微晶因各向异性在晶界处产生的残余应力,减少应力集中点,使裂纹在穿越晶界扩张时消耗更多能量,阻止裂纹的扩展,从而在碳纤维表面受到氧化刻蚀的同时限制碳纤维拉伸强度的损失量。此外,表面微晶尺寸的减小,使得晶界增多,处于碳纤维表面棱角和边缘位置的活性碳原子数也,就越多,这有利于提高纤维与树脂的粘结性,提高碳纤维复合材料的层间剪切强度。由表1和表2中数据可见,T-90及T-120的La值均小于T-0的,纤维在受氧化刻蚀的同时,本征拉伸强度的损失量均<10%,这与前人的研究相符[8.10-11]。但前人研究中也指出[10]:细晶化作用不能提高拉伸强度的原因是电化学改性会使碳纤维外层有序晶体层剥落,露出内部无序结构,引发新的缺陷。而且,T-150样品的拉伸强度损失量最小,但是其所对应的La值却最大。为此,对各实验样品的表面晶态结构进行拉曼光谱分析,分析其表面有序度的变化。

2.2碳纤维表面的Raman分析
图1为碳纤维的--级拉曼光谱,谱内明显存在两个峰: D峰和G峰。其中在约1350cm-1的D峰表示碳纤维结构中的无序结构,峰的强度与石墨晶格缺陷、边缘无序排列和低对称性的结构程度有关; G峰出现在1600 cm-1左右,该谱线是天然石墨所固有的,属于石墨晶格面内C-C键的伸缩振动。随着碳结构有序程度和石墨化程度的增高,D峰的强度逐渐减小,G峰的强度逐渐增大。因此,碳结构的有序度通常用代表无序结构的D峰与石墨结构的G峰的积分强度比值R(ID/IG)来进行表征[12-13]。
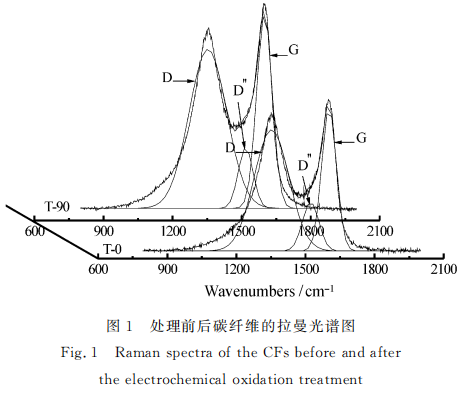
表3分别给出了各个样品按Gaussian曲线拟.合后的光谱参数。结合图1可以看出,改性后R值由T-0的2.02减小到T-90的1.91,再增加到T-120的2.01,而后又减小到T-150的1.90。这反映出碳纤维表面结构有序度随着电化学反应的加深,呈现先增大再减小,又增大的变化趋势。电化学改性后碳纤维表面有序度变化的差异源自氧化刻蚀程度的不同。表面处理中,氧化刻蚀作用在细晶化的同时率先去除碳纤维表面薄弱层、依附于表层的微小石墨片层和微晶与微晶之间的无定形结构,提高纤维表面的有序度。碳纤维边缘结构有序度以及晶体完整度的增加减少了缺陷存在的可能,有效地维持了纤维的拉伸强度[14-15]。与此同时,表面薄弱层及无定形结构的剥离,减少了碳纤维表面与树脂基体间的薄弱环节,有利于提高纤维与树脂的粘结性,提高碳纤维复合材料的力学性能。随后,由于内部无序结构的暴露,使得纤维表面的有序度减小。而T-150处纤维表面的有序度增加,可以理解为由于一些新生的无序结构被进一步的氧化刻蚀剥离所致,使纤维的拉伸强度和复合材料的层间剪切强度同时得以提高。
2.3 碳纤维表面的SEM分析
图2为碳纤维电化学氧化前后的表面SEM形貌。由低放大倍数的扫描电镜照片可以看出,碳纤

维表面呈现出许多窄且深浅不一的沿纤维轴向平行排列的沟槽,这是PAN基碳纤维生产过程中由其湿法纺丝原丝本身的带状微纤遗留下来的。比较表面处理前后的SEM照片,可以看出电化学改性对碳纤维表面宏观形貌的刻蚀效果并非完全像前人研究所述的表面沟槽会加宽加深。相反,由于沟槽或长棱的顶部会在电化学反应中汇集更多的电荷,产生电荷集中,率先成为电化学反应的主要场所,氧化刻蚀作用也率先在这些部位发生。因而,这些宏观的沟槽和长棱整体呈现的是变浅和均匀化的趋势,只有在改性程度进一-步加深后,这些沟槽和长棱才有可能因过度刻蚀而加宽加深。当加宽和加深的现象仅出现在局部区域时,会产生新的裂纹和缺陷,导致碳纤维拉伸强度损失。
由高放大倍数的扫描电镜照片可以看出,未改性碳纤维表面的沟槽呈颗粒状,具有类似龟裂的特征,经过电化学改性后,表面的颗粒变小或消失,微观表面趋向细腻光滑。而且这种表面结构的尺寸均在100 nm以下,在沿碳纤维轴向方向具有一定长度的连续性,并在未改性碳纤维上比较明显。该结构可能是碳化后表面沉积碳以表面微晶为基底沉积而成的,是一种表面弱层的组织形式;也有可能是表面微晶直接裸露形成。改性后,这种结构的尺寸明显减小,说明氧化刻蚀作用不仅剥离了碳纤维的表面弱层,同样对碳纤维表面晶体结构也有影响。
图3为碳纤维/树脂复合材料T-0及T-90样品的断面形貌。发现,未经表面处理的碳纤维复合材料断面凹凸不平,断面有大量纤维被拔出,出现较多的孔洞,说明纤维与树脂间的粘结性较差,导致其层间剪切强度低。表面处理后的碳纤维树脂基复合材料断面较为平整,基本在一个平面内;没有明显的单根纤维,孔洞较少,纤维与基体间没有缝隙,结合紧密,进一步说明阳极氧化提高了CFRP层间剪切强度。

由以上分析可见,碳纤维经过适度表面处理后其表面粗糙度降低,但其与树脂基体之间的粘结性却提高。在机械锚和作用降低的情况下,纤维与树脂基体之间的结合性依然得到提高,究其原因,应该是电化学氧化作用下,纤维表面化学活性得到提高,增强了纤维与树脂基体之间的化学键合作用,进而增强了纤维与树脂基体之间的粘合作用。
2.4碳纤维表面的XPS分析
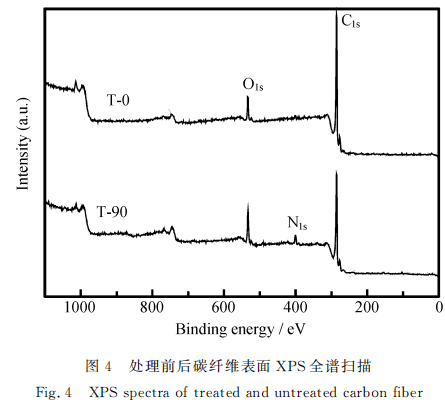
图4为电化学氧化处理前后碳纤维的全谱扫描,发现经过氧化处理后的碳纤维表面的Cis峰强度明显降低,Os峰、Ns峰有所增强。表4为电化学氧化后碳纤维表面各元素含量的变化,可以看出,随着氧化时间的增加,碳纤维表面含氧量呈现上升趋势;碳纤维表面含氮量也明显高于未改性碳纤维。随着反应程度的继续加深,氮元素含量有所波动,这是由于碳纤维表面层被刻蚀,导致含氮官能团随之被刻蚀剥离。此外,改性后的碳纤维表面均出现了硅元素,这可能是由于PAN原丝表面的含硅油剂渗人纤维内部,在碳化过程中被保留下来,在表面处理过程中随着碳纤维表面层的刻蚀剥离而露出表面产生。这种硅元素有可能在碳纤维内部聚集,阻碍石墨微晶在碳化过程中的成长,从而造成缺陷,当这些缺陷被暴露于碳纤维表面时,碳纤维的拉伸强度降低。结合碳纤维层间剪切强度,分析O/C和N/C发现,相比较氧元素的含量,氮元素含量与碳纤维复合材料的层间剪切强度关系更紧密,ILSS在T-90、T-120、和T-150的变化趋势与氮含量的变化相似。
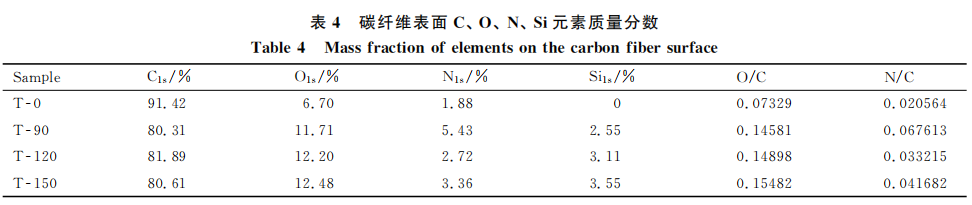
将C1s峰经XPEPEAK4.1进行分峰,计算碳纤维表面实际含氧官能团比例如表5所示。可见,电化学氧化后碳纤维表面羟基含量呈上升趋势,这是由于在电化学氧化的作用下,碳纤维表面活性碳原子吸附了碱性溶液中的OHT,并进一步反应生成C-OH,反应式如下:
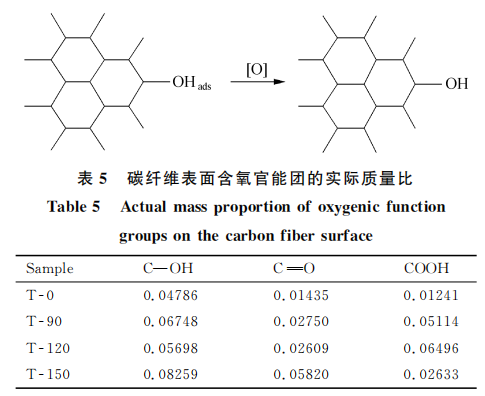
随着改性时间的变化,碳纤维中的羰基含量也逐渐增加,羰基可能是碳纤维表面边缘碳原子被电解出的活性氧直接氧化生成,但主要来源于C-OH的氧化,如下式:
碳纤维表面的羧基随着改性时间的增加呈现先增后减的趋势,由于未改性碳纤维表面本身所含的羧基很少,羧基含量的变化主要是电化学反应所致。在电化学处理时,阳极碳纤维上发生了如下反应[16]。
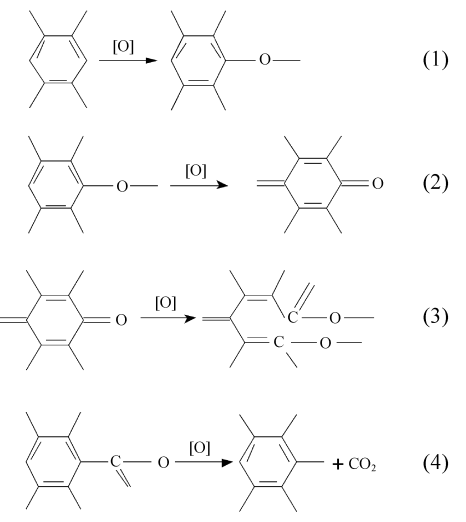
在氧化初期式(1)~式(3)反应占主导地位,同时式(4)的反应也在进行。可见随着氧化程度的加深,碳纤维表面的不饱和碳原子先后被氧化成羟基、羰基和羧基,而其表面原有的含氧官能团继续氧化。羧基在这类剧烈的放热反应中不稳定,极易释放出CO2,因此,随着反应的进行,式(4)成为主导反应[7]。每个羧基被氧化的位置会生成一个易被氧化的活性碳原子,这些活性碳原子随后被氧化成羟基和羰基,所以在T-150处碳纤维表面羟基和羰基的含量均很高,而羧基的含量降低。
由于羧基可以与环氧树脂基体发生化学反应,生成化学键合,有利于提高碳纤维复合材料的层间剪切强度。因此,在不损害碳纤维表面结构的情况下,羧基的含量与碳纤维复合材料的层间剪切强度有着直接的关联。然而在此试验中,碳纤维复合材料的层间剪切强度并未随羧基的增加而增加,也未随羧基含量的减少而减少。这说明碳纤维表面化学状态与其表面性能仍存在复杂的关系,是由多方因素(如:含氧官能团、含氮官能团、活性碳原子数量、晶体结构等)共同作用决定的。在150s处,虽然羧基含量与未改性纤维相近,但有别于未改性纤维的是其表面化学状态也随电化学改性发生了变化,首先羧基被氧化成CO2所遗留下的活性碳原子同样增加为碳纤维表面的化学活性点,其次,羟基和羰基含量的大幅度增加、含氮官能团的增加都为碳纤维与树脂基结构提供了化学键合的可能。但也需要注意的是,有研究认为[17]碳纤维进行表面改性时,羧基含量应尽量少。因为羧基在产生过程中,为使两个氧原子与碳原子键合,碳纤维表面石墨微晶的六元环必须断裂,破坏了石墨微晶的边缘部分,导致与羧基相连的炭层变脆。这样即使羧基与树脂粘结的很牢固,但易碎的炭层会分层,结果反而使CFRP的ILSS下降。这一.理论与120s改性时的效果正好吻合,因此,改性后羧基含量并非是越多越好,而是应控制在一个合适的范围内。
综上,碳纤维经表面处理后,碳纤维表面含氧官能团数目增多,其表面极性提高,利于极性环氧树脂对碳纤维表面的吸附和润湿,这样两者在界面上实现了分子接触;同时大分子通过内旋转运动,建立最合适的构象,达到吸附平衡,并通过吸附作用进行跨越界面的扩散,形成跨越界面的次价力或化学主价力的粘合键[18]。
在碳酸氢铵溶液中改性很容易在碳纤维表面生成含氮官能团,使碳纤维表面含氮量增加。这是因为,碳酸氢铵中的氨分子被碳纤维表面不饱和碳原子吸附,然后在电化学条件下生成氨基和亚氨基[19],反应历程如下:
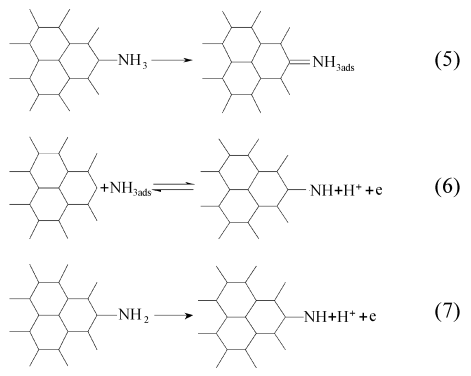
其中,在阳极发生电化学反应的氨分子主要来源于碳酸氢铵在水溶液中的水解以及铵根离子在阴极的还原。
图5为碳纤维表面NIs谱图。可以看出,碳纤维表面含有三类N官能团。一类在398.5eV附近的一N=,一类在400 eV附近的NH2(-NH),另一类是401. 5eV附近的氧化的氮,可能为一ONH4基团[8]。电化学改性后一N=官能团所占比重明显增加,说明碳纤维表面的确发生了氨基和亚氨基的生成反应。随着反应的进行,溶液中的氨分子不断地在碳纤维表面发生反应,生成含氨官能团,从而使铵根离子不断地在阴极还原,溶液中的铵根离子浓度不断下降。
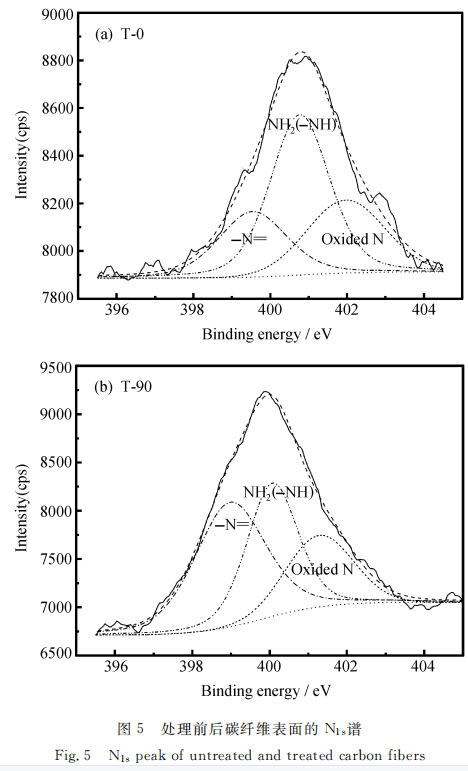
从电化学氧化后的各元素含量的变化来看(表4所示),碳纤维表面的氮元素含量的增加相当显著,但随着改性时间的增加氮的含量略有所下降。这也说明了电化学氧化过程中存在着两种作用,即氧化作用和刻蚀作用。氮元素在纤维表面先增加后又降低,正是刻蚀和氧化共同作用的结果。此外,若氮以吡啶结构的形式存在于碳中,当发生剧烈的氧化作用时,也有可能转化为含氧官能团,从而使氮含量降低,如下:
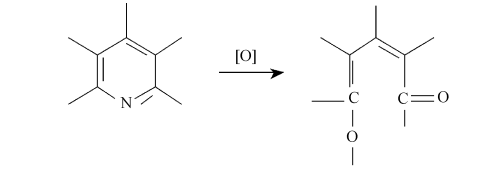
总之,XPS分析表明,碳纤维经电化学氧化处理后,表面引入了大量的含氧官能团和含氮官能团。由于这些官能团的增加,提高了碳纤维与环氧树脂的浸润性和反应性,有利于改善碳纤维/树脂复合材料的力学性能。
2.5 层进式物化双效模型
根据实验结果综合分析发现:电化学氧化处理对碳纤维本体同时具有物理作用(细晶化作用和氧化刻蚀作用)和化学作用(表面氧化作用)。初步推测电化学氧化法对碳纤维表面改性作用的时效性模型命名为“层进式物化双效"模型,认为在电化学改性中碳纤维外层的石墨微晶平面层在氧化的过程中被逐层刻蚀。根据该模型,实验中不同改性时间下的样品其层间剪切强度和拉伸强度变化过程可分为三个阶段(图6)。第一阶段:氧化反应首先发生在最表层的微小片层边界、乱层区域和无定形区域。使碳纤维表面含氧、含氮官能团增加,提高了碳纤维复合材料层间剪切强度;与此同时,刻蚀作用和细晶化作用使得碳纤维表面有序度增加、晶体尺寸减小,从而在纤维受到氧化刻蚀的同时较好的维持纤维的拉伸强度,该阶段末期时拉伸强度与层间剪切强度均达到理想状态。
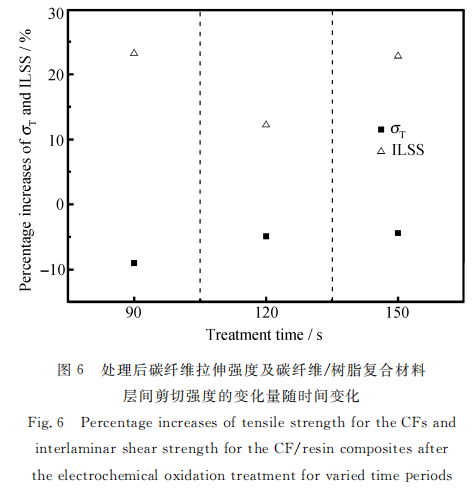
随着电解停留时间的增加,表面处理进入第二阶段:由于进一步的氧化刻蚀碳纤维表面的微小片层和无定形区域大部分被剥离,表面缺陷减少,结合细晶化效应,拉伸强度有所提高;但由于氧化活性点被刻蚀,表面含氮官能团含量大幅降低,碳纤维/树脂复合材料层间剪切强度有明显回落。随氧化刻蚀程度的进一步加深,表面处理进入第三阶段:此时,碳纤维表面的片层因深度刻蚀被破坏,再次出现微小片层,产生了新的氧化活性点,碳纤维/树脂复合材料层间剪切强度有所回升;新薄弱点边缘的尖锐部分在表面处理.作用下被钝化,有利于拉伸强度的提高,但由于过度的氧化刻蚀,拉伸强度并不会有较大回升。
3结论
(1)电化学表面处理对碳纤维具有细晶化作用和氧化刻蚀作用。细晶化作用有利于层间剪切强度和拉伸强度的提高。而适度的氧化刻蚀作用不仅可以除去碳纤维表面薄弱层、微小石墨片层和微晶与微晶之间的无定形结构,减少碳纤维表面与树脂基体间的薄弱环节,有利于提高碳纤维/树脂复合材料层间剪切强度;而且可以增加碳纤维边缘结构有序度,有效发挥细晶化作用对拉伸强度的影响,降低碳纤维本征拉伸强度的损失。
(2)碳纤维经电化学氧化改性后,表面含碳量降低10%~12%,氧含量提高75%~86%,氮含量提高0.5~2倍。改性后碳纤维表面羟基和羰基明显增加,羧基在短时间改性时含量呈增加状态,而随着羧基被进一步氧化为CO2,其含量明显下降。含氧、含氮官能团的增加使得复合材料的层间剪切强度增加,但同时发现,当羧基含量下降时也并不影响复合材料的层间剪切性能。碳纤维/树脂复合材料的层间剪切性能是由多方面因素如:表面形貌、含氧官能团、含氮官能团、活性碳原子数量、晶体结构等共同作用决定的,这些因素之间也会有互补作用。
(3)在适当的处理条件下,随着反应程度加深,碳纤维拉伸强度呈现逐渐上升的趋势,复合材料层间剪切强度呈现先下降再上升的变化趋势,表面电化学氧化处理作用为逐层氧化刻蚀与官能团变化的复合作用过程。适度控制碳纤维结构变化,可以在提高层间剪切强度的同时降低拉伸强度的损失量。
参考文献:
[1] 贺福.碳纤维及其应用技术[M].北京:化学工业出版社,2004: 233-256.
[2]刘杰,郭云霞,梁节英.碳纤维表面电化学氧化的研究[J]. 化工进展,2004, 23(3): 282-285
.Liu Jie, Guo Yunxia, Liang Jieying. Study on electrochemical loxidation of carbon fibers surface [J]. Chemical Industry and Engineering Progress,2004, 23(3): 282-285.
[3]朱民,龚真萍.粘胶基碳布电化学氧化表面处理的研究[J]. 华东师范大学学报:自然科学版,1998(2): 55-60.
Zhu Min, Gong Zhenping. A study of electrochemical oxidation surface treatment on rayon- based carbon fabrics [J] .Journal of East China Normal University: Natural Science ,1998(2): 55-60.
[4] He H, WangJ, LiK, WangJ, Gu J. Mixed resin and carbon fibres surface treatment for preparation of carbon fibres composites with good interfacial bonding strength[J].Materials & Design, 2010, 31(10): 4631-4637.
[5] ZhangG,Sun s,Yang D,et al. The surface analytical characterization of carbon fibers functionalized by H2 SO4/HNO3 treatment [J]. Carbon, 2008, 46: 196-205.
[6] Montes- Moran M A,Gauthier W, Martinez - Alonso A,TasconJ M D, et al. Mechanical properties of high- strength carbon fibres. Validation of an end-effect model for describingexperimental data [J]. Carbon, 2004, 42(7): 1275-1278.
[7]郭云霞,刘杰,梁节英.电化学改性PAN基碳纤维表面及其机理探析[J].无机材料学报,2009,24(4): 853-857.
Guo Yunxia, Liu Jie, Liang Jieying. Modification mechanism of the surface - treated PAN - based carbonfiber byelectrochemical oxidation [J]. Journal of Inorganic Materials,
2009,24(4): 853-857.
[8] 刘鸿鹏,吕春祥,李永红,等.电化学表面处理PAN基碳纤维的表面性能研究[J].新型炭材料,2005,20(1): 39-44.
Liu Hongpeng, Li Chunxiang, Li Yonghong, et al. Surface properties of electrochemically oxidized PAN - based carbon fibers [J]. New Carbon Materials, 2005, 20(1): 39-44.
[9] Mittal J, Bahl OP, Mathur R B. Single step carboniza tion and graphitization of highly stabilized PAN fibers [J]. Carbon,1997,35(8): 1196-1197.
[10]郭云霞,刘杰,梁节英,电化学改性对PAN基碳纤维表面状态的影响[J].复合材料学报,2005, 22(3): 49-54.
Guo Yunxia, Liu Jie,Liang Jieying. Effect of electrochemical modification on the surface state of PAN- based carbon fibers[J]. Acta Materiae Compositae Sinica,2005, 22(3): 49-54.
[11] Papirer E, Guyon E. Contribution to the study of the surface groups on carbons [J]. Carbon, 1978, 16(4): 127-133.
[12]HoKKC,,Lee A F, I . amorinierea S. Continuous atmospherie plasma fluorination of carbon fibres [ J ]. Composites: Part A, 2008, 39: 364-373.
[13]李东风,王浩静,王心葵. PAN基碳纤维在石墨化过程中的拉曼光谱[J]. 光谱学与光谱分析,2007, 27(11): 2249-2253.
Li Dongfeng,Wang Haojing, Wang Xinkui. Raman spectra of PAN - based carbon fibers during graphitization [ J ].Spectroscopy and Spectral Analysis, 2007, 27 (11): 2249 -2253.
[14]贺福.用拉曼光谱研究碳纤维的结构[J]. 高科技纤维与应用,2005, 30(6): 20-25.He Fu. Raman spectroscopy studies on structure of carbon fibres [J]. Hi- Tech Fiber & Application, 2005, 30(6): 20-25.
[15] Li D, Wang H, HeF, Wang X K. Structure and properties of T300 and T700 carbon fibers[ J]. New Carbon Materials,2007,22: 59-64.
[ 16] Bansal R,Donnet J,Stoeckli H,Active carbon[ M]. New York: NY, Marcel Dekker, 1998.
[17] Shimizu K,Nakahara M. Interfacial debonding strength between the edge surfaces of pyrolytic graphite and epoxy resins [J]. Journal of Material Science, 1992, 27(22): 6134-6140.
[18]张开.高分子界面科学[M].天津:天津大学出版社,1997.
[19]房宽峻.黏胶基碳纤维阳极氧化机理与工艺研究[D].上海:中国纺织大学,1993.

微信公众号
